Search
The Kids Research Institute Australia spin-off company, Respirion, received $20 million in funding to develop a promising new therapy.

The family of two girls with cystic fibrosis are hopeful after The Kids Research Institute Australia spin-off company, Respirion, receives $20 million in funding to develop a promising new therapy.

A The Kids Research Institute Australia spin-off company has received $20 million from the Medical Research Commercialisation Fund to develop a promising new therapy for the treatment of Cystic Fibrosis.

The Kids researchers are pioneering an exciting new approach to clinical trials, which aims to fast-track the best treatments for people with rare and complex diseases.

The Kids Research Institute Australia researchers have been awarded more than $8 million in prestigious project grants from the NHMRC.

PRAGMA-CF, a new way of measuring early lung disease in young kids with cystic fibrosis is changing the way we detect and treat CF.
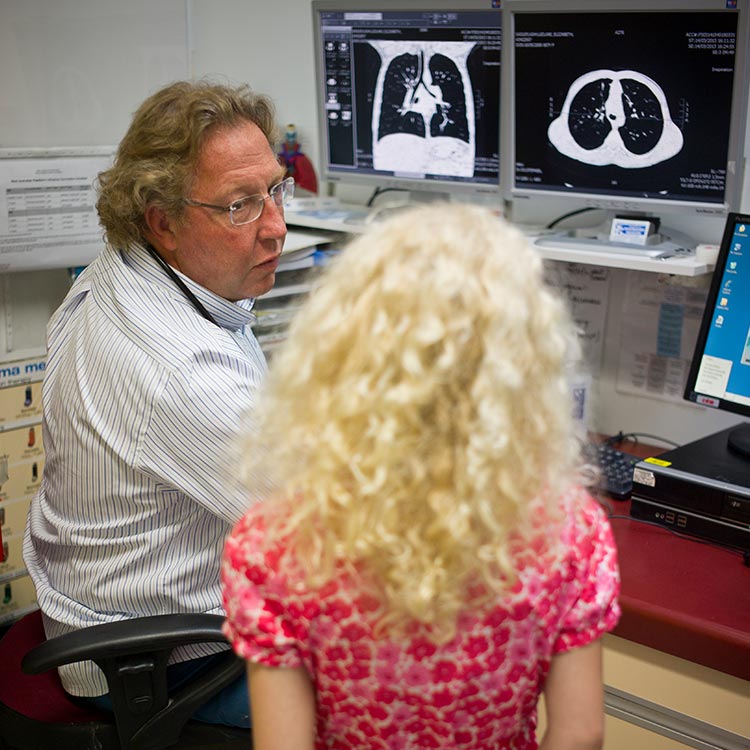
Early intervention is being touted as the key to preventing lung damage in children with cystic fibrosis.

When Ingrid Laing was born, the outlook for kids with cystic fibrosis was bleak. Her parents were told she might make it to 20 if she was lucky.

We are looking for 6 new members to join our Child and Adolescent Cystic Fibrosis Consumer Reference Group of WA

That's why Melissa has signed up her four year old healthy son Odin for a study at Perth's The Kids Research Institute Australia that will help kids with cystic fibrosis.